
Protein Engineering
FoldX
FoldX provide a fast and quantitative estimation of the importance of the interactions contributing to the stability of proteins and protein complexes. The predictive power of FOLDEF has been tested on a very large set of point mutants (1088 mutants) spanning most of the structural environments found in proteins. FoldX uses a full atomic description of the structure of the proteins. The different energy terms taken into account in FoldX have been weighted using empirical data obtained from protein engineering experiments.
FoldX was created in the Serrano lab at the Heidelberg Laboratory of the EMBL by Raphael Guerois, Jens Nielsen, Jesper Ferkinghoff-Borg, Joost Schymkowitz, Frederic Rousseau, Francois Stricher and Luis Serrano.
DnaX
People involved: Hector Climente, Javier Delgado
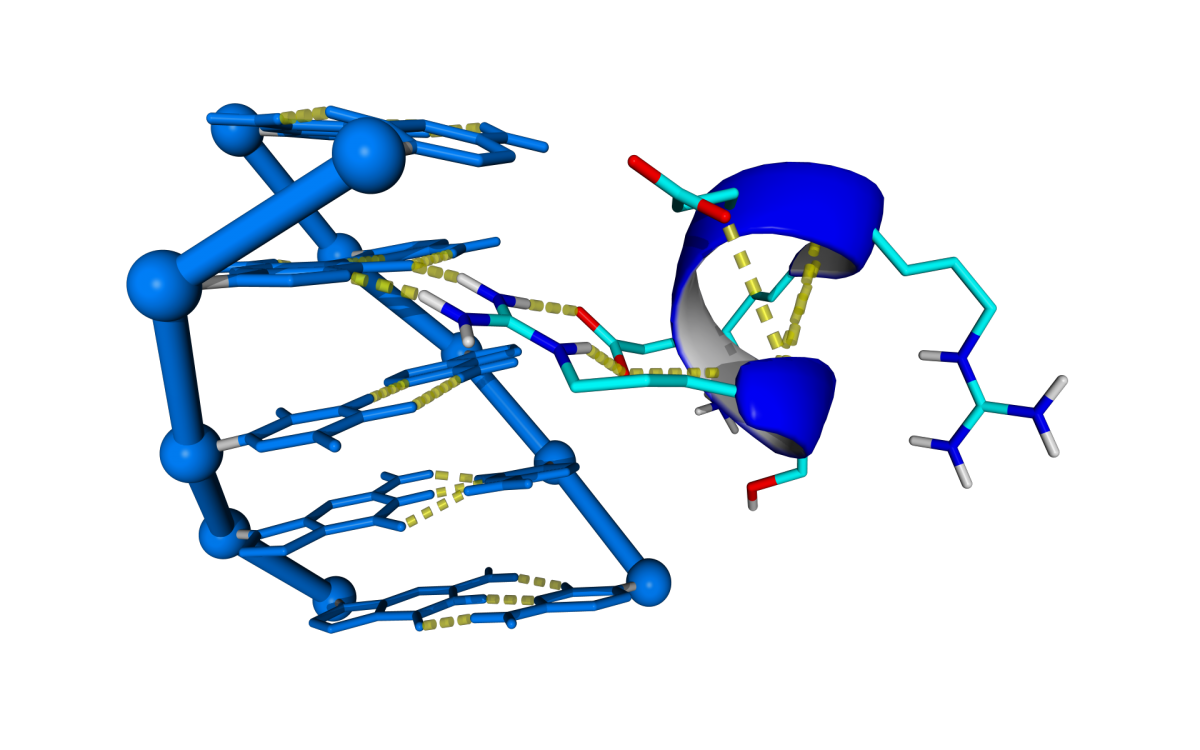 The number of Protein-DNA crystal structures in a complex deposited at PDB are exponentially increasing its number during the last 25 years. The project aims to predict DNA docking into protein by using the structural information stored in these crystal structures. The approach for DNA binding prediction we use is based in the assumption that Prot-Dna interactions occurs in a limited number of conformations. With that purpose in mind we built a repository of Prot-Dna interactions by digesting In Silico more than 2500 Prot-Dna crystal structures classifying and capturing the structural information represented by those interactions.
The number of Protein-DNA crystal structures in a complex deposited at PDB are exponentially increasing its number during the last 25 years. The project aims to predict DNA docking into protein by using the structural information stored in these crystal structures. The approach for DNA binding prediction we use is based in the assumption that Prot-Dna interactions occurs in a limited number of conformations. With that purpose in mind we built a repository of Prot-Dna interactions by digesting In Silico more than 2500 Prot-Dna crystal structures classifying and capturing the structural information represented by those interactions.
Modeling specificity of peptide recognition domain families
People involved: Erik Verschueren, Peter Vanhee
Keywords: protein-peptide interactions, signaling pathways, systems biology, protein structure
Modular protein interaction domains form the cornerstone of eukaryotic signaling pathways. These domain families have adapted themselves through evolution to selectively recognize short linear polypeptide motifs in other proteins.
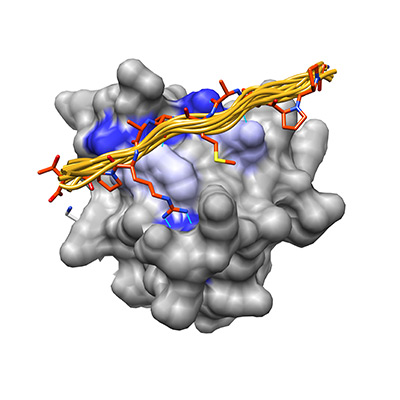 In my work I study how two peptide recognition domains, the PDZ domain and SH3 domain, have optimized their specificity landscape to minimize cross-reactivity while retaining the core binding motifs that characterize them as a family.
In my work I study how two peptide recognition domains, the PDZ domain and SH3 domain, have optimized their specificity landscape to minimize cross-reactivity while retaining the core binding motifs that characterize them as a family.
Recent developments in high-throughput binding assays have uncovered the complexity of these recognition domains, making these methods the tool of choice for specificity studies. Nevertheless, we demonstrate that three-dimensional structures of protein-peptide interactions are a valuable, complementary resource to truly understand the subtle interface changes that enable these domains to selectively bind their ligands. Unfortunately, structural coverage of peptide recognition modules in complex with their ligands is low and computational algorithms have not fully tackled the challenges of peptide structure prediction yet.
We propose the use of large libraries of polypeptide fragments to improve peptide prediction methods. We recently found that the majority of protein-peptide architectures can be represented as interactions between fragments in single, unrelated structures and developed an algorithm that predicts the structure of peptides bound to their partner using these interaction patterns. We project that fragment-based structure prediction methods hold great promise to further explore, understand the complexity and ultimately engineer protein-peptide interactions in signaling pathways.
Protein-DNA Interaction
People involved: Andreu Alibés, Alejandro Nadra, François Stricher
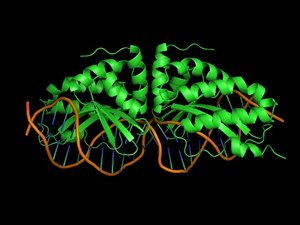 Binding of proteins to nucleic acids is crucial for the regulation and fulfillment of many biological processes. Yet, there is no reliable method that can predict protein-DNA interaction affinity and specificity. Nowadays, rational design of proteins is in many aspects a routine procedure provided that a good structural template is available.
Binding of proteins to nucleic acids is crucial for the regulation and fulfillment of many biological processes. Yet, there is no reliable method that can predict protein-DNA interaction affinity and specificity. Nowadays, rational design of proteins is in many aspects a routine procedure provided that a good structural template is available.
However, protein-DNA interactions are still at the frontier of protein design. This is mainly due to the fact that water mediates in many cases protein-DNA interactions as well as the lack of experimental data that will allow proper calibration of the force fields.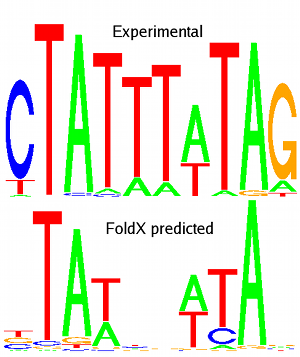
Our aim is to predict affinities and specificities based on structural information and to be able to design new specificities and/or modify affinities at will. The theoretical analysis will be done mainly by an improved FoldX version capable of dealing with protein-DNA complexes.
Pictures: MEF2A experimental (from JASPAR database) and predicted logos
Protein-Protein and Protein-Peptide
Understanding and redesign of Protein-Protein interactions
People involved: Anne Campagna, Christina Kiel, Almer Van der Sloot
Protein-protein interactions play a central role in cellular signaling events by transducing signals within the cell and between cells. These interactions are often mediated by evolutionary and structurally well conserved domains and modules. Using software tools (e.g. FoldX) developed in our group we aim to develop a deeper understanding of various signaling networks from a structural perspective. Topics such as which structural features determine the specificity within these networks and of its individual domains and modules are addressed. In addition to predictions, considerable effort is invested in the experimental validation of these predictions. Successful predictions have been carried out for Ras-effector interactions, and interactions of PDZ and SH3 domains with their target peptides.
Further, we use structural information in order to increase the information of large scale experimental and bioinformatics-based protein-protein interaction networks. We are addressing the following questions: If several proteins are found experimentally in one complex, which proteins can make direct interactions? Which of the protein interactions can occur at the same time?
In diseases like cancer, normal signaling networks are frequently perturbed. Restoring the normal physiological signaling networks, switching off disturbed networks, or triggering alternative signaling cascades can be a useful strategy in treating the disease. Using the protein design algorithm FoldX, we design proteins and peptides that either switch-on or switch-off a signaling route with high specificity and that can be used as potential therapeutic. Signaling networks triggered by TNF-family receptors are one of our model systems, by blocking cell proliferation signals or stimulating cell-suicide (apoptosis) signals with specifically designed proteins or peptides we seek to develop new therapeutic strategies to combat cancer.
Protein complex dynamics
People involved: Tony Ferrar, Tobias Maier
It became evident over the last years, that proteins predominantly do not act as single molecules, but rather perform their functions in conjunction with other proteins, as complexes. Large scale studies in different organisms showed that the organization of the proteome in protein complexes is ubiquitous, and applies to many cellular processes.
We study the dynamics of identified protein complexes changing their composition along the cell cycle in vivo. Isolation of protein complexes by affinity purification methods, coupled to current mass spectrometric methods are used to identify and quantify the composition of analyzed protein complexes.