
Cell Signaling
Quantitative Systems Analyses of Mammalian Signaling
In recent years, high-throughput discovery of macromolecular protein structures and complexes has played a major role in advancing a more systems-oriented view of protein interaction and signaling networks. In the signaling sub-team, we employ structural information or structure-based protein design to successfully implement synthetic signaling circuits or for rewiring signaling flow. Combining structural biology with engineered cell signaling modules--using additional information from quantitative biochemistry and proteomics, gene evolution, and mathematical modeling--can provide insight into signaling modules and the general design principles of cell signaling. This will improve our understanding of cell- and tissue-type-specific signal transduction. Integrating the quantitative effects of disease mutations into these systems may provide a basis for elucidating the molecular mechanisms of diseases.
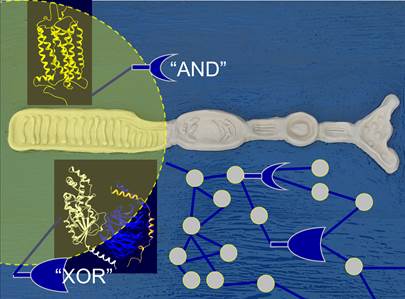
The role of protein competition in signaling networks
The mechanisms of context-specific differences in signal transduction among cell types are not fully understood. One model posits that differences in protein concentrations could change signaling outputs because of competition for common binding partners at critical network branch points. We analyzed competing protein interactions in the ErbB signaling and in the rhodopsin network (collaboration with Marius Ueffing, Tuebingen) (1). We created a high-confidence protein interaction networks and identified domains and motifs mediating interactions and their affinities. Using three-dimensional protein structures and modelling (2), the network was dissected into compatible (AND) and mutually exclusive (XOR) protein interactions. Using quantitative information about protein abundances, our model uncovered a high degree of competition at XOR nodes. We are in the process of characterizing different network motifs experimentally. So far our results indicate that variation in the abundance of proteins that compete for binding to XOR network motifs could contribute to cell type-specific signaling plasticity. This project is related to the EU projects PRIMES and PROSPECTS.
The role of kinetic constants (association and dissociation constants) for signal transmission
Recently, we have designed mutations in the Ras-CRAF interface with compensating changes in association and dissociation rates, but otherwise similar affinity (3). In RK13 cells, these mutants affected downstream signaling, with changes in Ras-c-Raf association rates having a greater effect on MAPK signaling than did similar changes in dissociation rates. In marked contrast, the mutants had little or no effect on signaling in human embryonic kidney (HEK) 293 cells. These two cell lines also showed distinct patterns of EGF-dependent ERK phosphorylation and signaling: ERK activation and signaling were transient in HEK293 cells and sustained in RK13 cells, with the difference resulting from the lack of negative feedback from ERK to Sos (Son of Sevenless) in the latter. Computer simulation revealed that, in the presence of negative feedback, changes in the rate of Ras-c-Raf binding have little effect on ERK activation. Thus, EGF-MAPK activation kinetics and feedback regulation is cell type specific and depends on the network topology.
We have employed a similar concept to the Hog1 signaling in yeast (in collaboration with Francesc Posas, Barcelona and Edda Klipp/ Berlin) . We investigated the role of kinetic rate constants of a key upstream complex in the Hog1 pathway, namely of the response regulator domain (RR) of Sln1 and the histidine-containing phosphorelay protein Ypd1. Using X-ray structural information and the protein design algorithm FoldX, we engineered amino acid mutations with only moderate perturbations in complex affinity by increasing kon through electrostatic steering and increasing koff through mutation of hot-spot interface residues. While computer modelling suggest that faster association and dissociation should improve signal transmission, preliminary experiments suggest that the opposide is true: at high osmotic stress, when also the intracellular ionic strength in yeast cells increases to ~ 0.6M, the kinetically optimized mutations show a phenotype, as the internal ionic strength weakens the mutant complexes by screening electrostatic interactions and decreasing kon. In contrast, the wild type complex is unaffected by the increase in ionic strength. Indeed, our results suggest a functionally important hydrophobic interface conservation of the Sln1-Ypd1 complex. This project is related to the EU project UNICELLSYS.
Cell general and cell type-specific proteins in the ErbB signaling network: towards a complete quantitative description of a signaling pathway
Relating protein concentration to cell type-specific responses is one of the remaining challenges for obtaining a quantitative systems level understanding of mammalian signaling. In this project, we used mass spectrometry- and antibody-based quantitative proteomic approaches to measure protein abundances for 75% of a hand curated reconstructed ErbB network of 198 proteins, in two established cell types (HEK293 and MCF-7) and in primary keratinocyte cells. Comparison with other quantitative studies allowed building a set of ErbB network proteins expressed in all cells, and another which are cell specific and could impart specific properties to the network. As a proof-of-concept of the importance of protein concentration, we generated a small simplified mathematical model encompassing ligand binding, followed by receptor dimerization, activation and degradation. The model predicts ErbB phosphorylation in HEK293, MCF-7, and keratinocyte cells simply by incorporating cell type-specific ErbB1, ErbB2 and caveolin-1 abundances, but otherwise contains similar rate constants. Altogether, the data provide a resource for protein abundances and localization. We are now extending the model to a much larger type specific computational mathematical model, which should predict the effect of downstream signaling merely based on protein abundances, but otherwise similar rate constants. This project is related to the EU project PRIMES.
Cell type specific signaling networks: towards a global analysis of cell signaling pathways
In this project, we related protein expression levels in 11 human cell lines and evolutionary conservation to 10 general cellular pathways. We find that proteins in signaling-related pathways are less conserved and have higher protein abundance variability than typical housekeeping associated pathways. We observe strong differences in protein abundance variability and conservation between different functional and topological classes within human signaling pathways: proteins associated with the downstream transmission of the signal tend to have a higher conservation and a moderate to low protein abundance variability over cell types. These proteins preferentially link to highly variably and less conserved receptors which likely modulate cell type specific activity of the signaling pathways. The differences in expression and conservation between signaling proteins are accompanied by different tendencies to acquire mutations: signaling proteins with somatic mutations tend to have a higher conservation and lower protein abundance variability then proteins with germline mutations. We propose that the modulation of the activity of static signal transmission machinery by receptor abundance is a main determinant of tissue-specificity and common mechanism in human signaling. We are planning experimental functional assays to validate these hypotheses. This project is related to the EU projects PRIMES and PROSPECTS .
Alternative splicing in the ErbB signaling network
Our structural network with domain-domain and domain-linear motifs annotated suggests a large number of splice variants that may affect protein interactions. We are in the process of analysis the role of splice variants systematically on one complete signaling pathway. We are in the process of measuring the abundance of splice variants in different cell lines using selected reaction monitoring (SRM) mass spectrometry. We are also developing a mathematical tool that can be used to quantify the abundance of splice variants from mass spectrometry experiments, by incorporating shared and unique peptides for splice variants. This project is related to the EU project PRIMES.